
审评提示丨有源医疗器械产品注册审评提示




2020年以来,湖北省药监局在全国首创审评提示和审评共识服务机制,让企业与审评部门沟通更规范、更高效。2021年6月21日,发布《关于建立“两品一械”审评提示和审评共识服务机制的通告》,将服务机制进一步定型化。审评提示和审评共识服务机制实施以来,湖北省药监局发布了多期提示和共识,促进企业产品上市更顺畅。2022年,湖北药监继续推出《审评提示》专栏,敬请关注。
有源医疗器械产品注册审评提示
2022年第8期
为指导企业注册申报,针对我省第二类有源医疗器械注册过程中的高频问题,发出以下审评提示:
医用光学内窥镜、激光光纤是否需要进行电磁兼容检验?
答复:如果医用光学内窥镜、激光光纤内部不包含电子元器件,仅仅包含光学元件,则不需要进行电磁兼容检测。如果内部含有电子元器件(如RFID识别装置等),则需要进行电磁兼容检测。
医疗器械独立软件的产品名称和产品描述与免于临床评价医疗器械目录中产品表述一致,但采用了全新算法,产品是否仍然可豁免临床评价?
答复:独立软件若采用了全新算法需按照《医疗器械临床评价技术指导原则》的要求进行临床评价。
医用软件产品的有效期如何确定?
答复:独立软件的使用期限通过商业因素予以确定,应在产品设计开发过程中,通过对比同类产品、市场调研、产品预期使用年限等方式进行合理定义。软件组件的使用期限与所属医疗器械相同,无需单独体现。专用型独立软件视为软件组件的使用期限要求与独立软件相同,在所属医疗器械使用期限研究资料中体现。
关于补充检验是否应在原检验机构进行检验?
答复:申请注册提交的医疗器械检验报告为委托有资质的医疗器械检验机构出具的检验报告,注册审查时提出补充检验要求的,补充检验可以委托其他有资质的医疗器械检验机构进行检验,也可以由医疗器械注册申请人进行自检,但应保证检验样品的一致性。
注册申请人应如何提交自检报告?
答复:注册申请人提交的自检报告要求应是基于申报产品技术要求全项目的检验报告。格式应符合《医疗器械注册自检管理规定》“附件1《医疗器械注册自检报告》(模板)”的要求。部分项目涉及委托检验的,注册申请人应当对受托方出具的报告进行汇总,结合注册申请人自行完成的检验项目,形成完整的自检报告,除在备注栏注明受托的检验机构外,还应在自检报告后附上委托检验报告原件。
来源:湖北药监
(责任编辑 龚晓艺)


 融媒体平台建设服务
融媒体平台建设服务 长江云新时代文明实践平台
长江云新时代文明实践平台

 大数据舆情中心
大数据舆情中心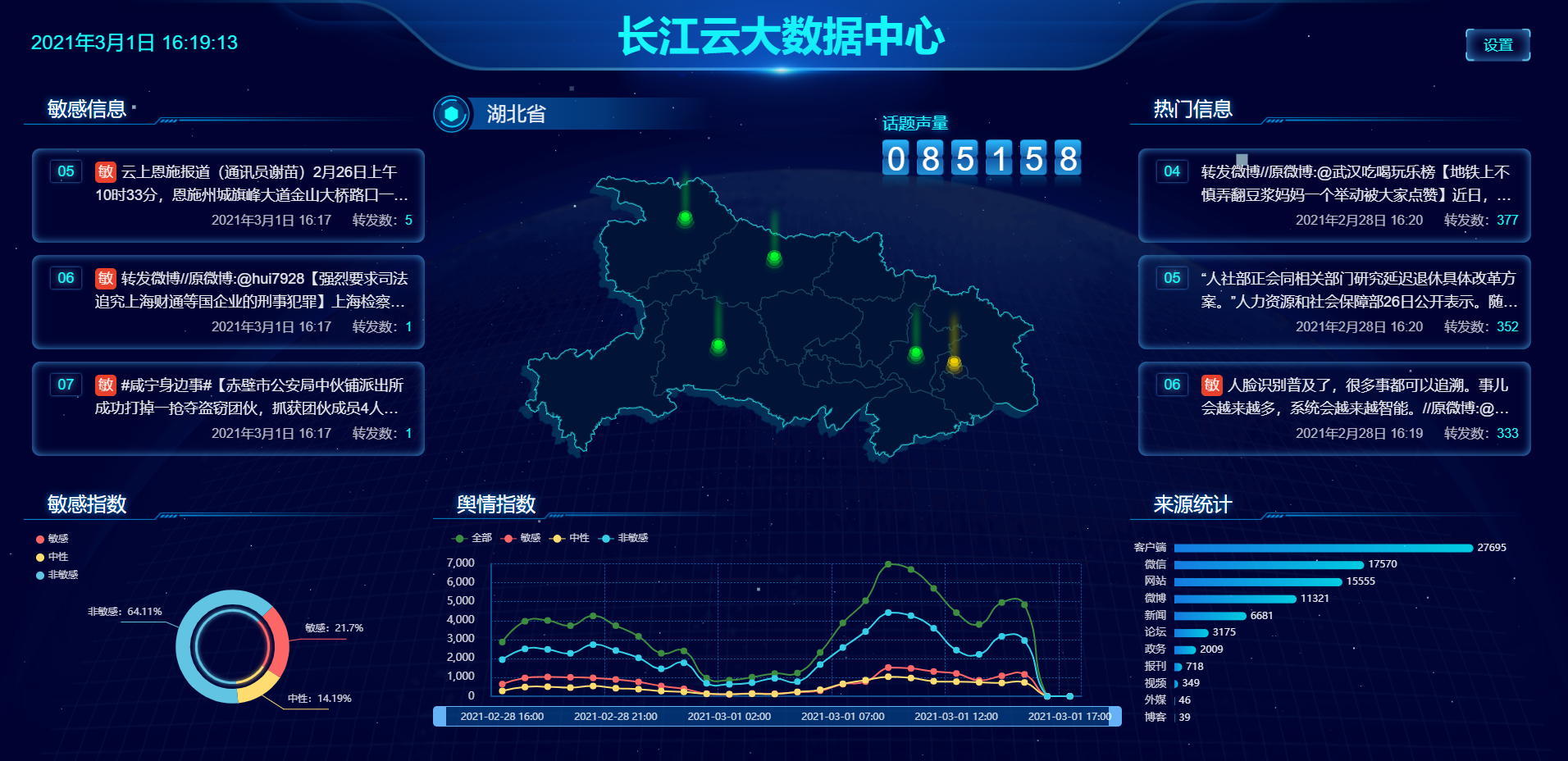


{kind=link}
{kind=link}